How does ALS Destroy the Human Body?
Amyotrophic lateral sclerosis or Lou Gehrig's disease is a neurodegenerative disorder with various causes.
The term motor neurone disease (MND) is sometimes used interchangeably with ALS, while others use it to refers to a group of similar conditions that include ALS.
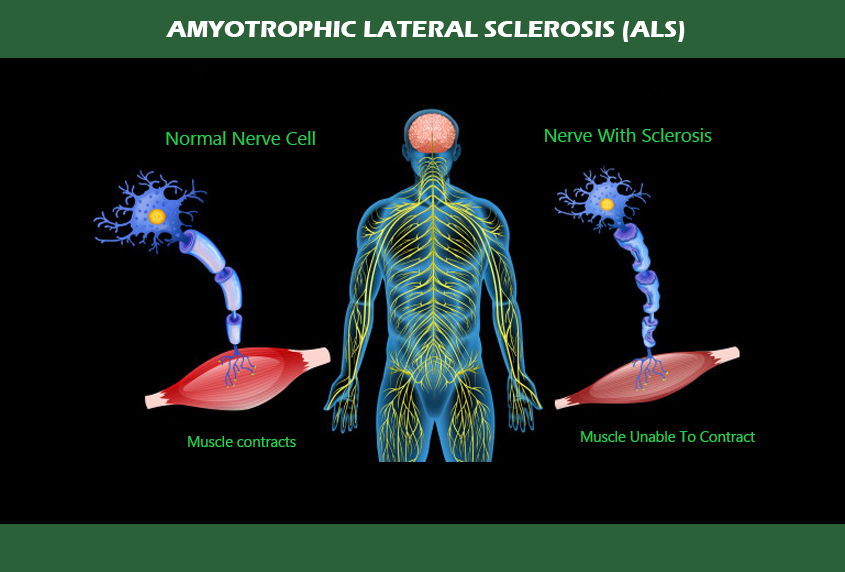
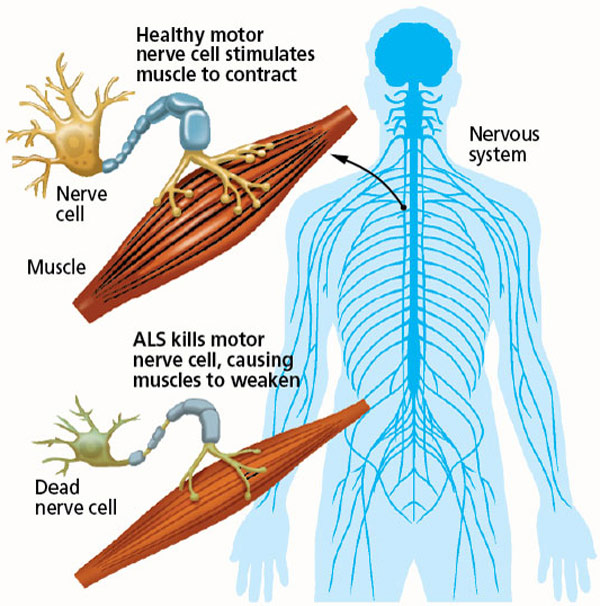
ALS is characterised by muscle spasticity, rapidly progressive weakness due to muscle wasting. This results in difficulty speaking, swallowing, and breathing.
The disease usually starts around the age of 60, except in cases that are directly inherited when the usual age of onset is around 50.
About 5 to 10% of cases are directly inherited from a person's parents. ALS is the most common of the five motor neuron diseases.
The average survival from onset to death is three to four years. Only 4% survive longer than 10 years, although rare cases survive 50 years or more. Most die from respiratory failure.
Signs and symptoms
The disorder causes muscle weakness and atrophy throughout the body due to the degeneration of the upper and lower motor neurons.
Individuals affected by the disorder may ultimately lose the ability to initiate and control all voluntary movement, although bladder and bowel function and the muscles responsible for eye movement are usually spared until the final stages of the disorder.
Cognitive function is generally spared for most people, although some (about 5%) also develop frontotemporal dementia.
Initial symptoms
The earliest symptoms of ALS are typically obvious weakness and/or muscle atrophy.
Other presenting symptoms include: trouble swallowing, cramping, or stiffness of affected muscles, muscle weakness affecting an arm or a leg, and/or slurred and nasal speech.
The parts of the body affected by early symptoms of ALS depend on which motor neurons in the body are damaged first.
About 75% of people contracting the disorder experience "limb onset" ALS, i.e., first symptoms in the arms or legs.
People with the leg onset form may experience awkwardness when walking or running or notice that they are tripping or stumbling, often with a "dropped foot" which drags gently along the ground.
Arm-onset people may experience difficulty with tasks requiring manual dexterity such as buttoning a shirt, writing, or turning a key in a lock.
Occasionally, the symptoms remain confined to one limb for a long period of time or for the whole length of the illness. This is known as monomelic amyotrophy.
About 25% of cases are "bulbar onset" ALS. These people first notice difficulty speaking clearly or swallowing. Speech may become slurred, nasal in character, or quieter. Other symptoms include difficulty swallowing and loss of tongue mobility.
A smaller proportion of people experience "respiratory onset" ALS, where the intercostal muscles that support breathing are affected first.
Over time, people experience increasing difficulty moving, swallowing (dysphagia), and speaking or forming words (dysarthria).
Symptoms of upper motor neuron involvement include tight and stiff muscles (spasticity) and exaggerated reflexes (hyperreflexia) including an overactive gag reflex.
An abnormal reflex commonly called Babinski's sign also indicates upper motor neuron damage.
Symptoms of lower motor neuron degeneration include muscle weakness and atrophy, muscle cramps, and fleeting twitches of muscles that can be seen under the skin (fasciculations).
Progression
Although the order and rate of symptoms varies from person to person, eventually most people are not able to walk or use their hands and arms. They also lose the ability to speak and swallow food, while most end up on a portable ventilator, called a BiPAP.
Disorder progression tends to be slower in patients who are younger than 40 at onset, are mildly obese, have disorder restricted primarily to one limb, and those with primarily upper motor neuron symptoms.
Conversely, progression is faster and prognosis poorer in patients with bulbar-onset disorder, respiratory-onset disorder, and fronto-temporal dementia.
Late stages
Although respiratory support can ease problems with breathing and prolong survival, it does not affect the progression of ALS.
Most people with ALS die from respiratory failure, usually within three to five years from the onset of symptoms. The median survival time from onset to death is around 39 months, and only 4% survive longer than 10 years.
Difficulty in chewing and swallowing makes eating very difficult and increases the risk of choking or of aspirating food into the lungs.
In later stages of the disorder, aspiration pneumonia can develop, and maintaining a healthy weight can become a significant problem that may require the insertion of a feeding tube.
As the diaphragm and intercostal muscles of the rib cage that support breathing weaken, measures of lung function such as vital capacity and inspiratory pressure diminish.
In respiratory onset ALS, this may occur before significant limb weakness is apparent. Most people with ALS die of respiratory failure or pneumonia.
In late stages, the oculomotor nerve that controls the movements of the eye, can be affected as can the extraocular muscles.
The eye movements remain unaffected largely until the later stages due to differences in the extraocular muscles compared to the skeletal muscles that are initially and readily affected.
Diagnosis
No test can provide a definite diagnosis of ALS, although the presence of upper and lower motor neuron signs in a single limb is strongly suggestive.
Instead, the diagnosis of ALS is primarily based on the symptoms and signs the physician observes in the patient and a series of tests to rule out other diseases.
Physicians obtain the patient's full medical history and usually conduct a neurologic examination at regular intervals to assess whether symptoms such as muscle weakness, atrophy of muscles, hyperreflexia, and spasticity are getting progressively worse.
Because symptoms of ALS can be similar to those of a wide variety of other, more treatable diseases or disorders, appropriate tests must be conducted to exclude the possibility of other conditions.
One of these tests is electromyography (EMG), a special recording technique that detects electrical activity in muscles.
Certain EMG findings can support the diagnosis of ALS. Another common test measures nerve conduction velocity (NCV).
Specific abnormalities in the NCV results may suggest, for example, that the patient has a form of peripheral neuropathy (damage to peripheral nerves) or myopathy (muscle disease) rather than ALS.
The physician may order magnetic resonance imaging (MRI), a noninvasive procedure that uses a magnetic field and radio waves to take detailed images of the brain and spinal cord.
Although these MRI scans are often normal in patients with ALS, they can reveal evidence of other problems that may be causing the symptoms, such as a spinal cord tumor, multiple sclerosis, a herniated disk in the neck, syringomyelia, or cervical spondylosis.
Other treatments for ALS are designed to relieve symptoms and improve the quality of life for patients.
This supportive care is best provided by multidisciplinary teams of health care professionals working with patients and caregivers to keep patients as mobile and comfortable as possible.
Medications
Medications may be used to help reduce fatigue, ease muscle cramps, control spasticity, and reduce excess saliva and phlegm.
Drugs also are available to help patients with pain, depression, sleep disturbances, dysphagia, and constipation.
Physical therapists and occupational therapists play a large role in rehabilitation for individuals with ALS.
Specifically, physical and occupational therapists can set goals and promote benefits for individuals with ALS by delaying loss of strength, maintaining endurance, limiting pain, preventing complications, and promoting functional independence.
When the muscles that assist in breathing weaken, use of ventilatory assistance (intermittent positive pressure ventilation (IPPV), bilevel positive airway pressure (BiPAP), or biphasic cuirass ventilation (BCV) may be used to aid breathing.
Such devices artificially inflate the person's lungs from various external sources that are applied directly to the face or body.
When muscles are no longer able to maintain oxygen and carbon dioxide levels, these devices may be used full-time.
© COPYRIGHT 2019-2020 ALL RIGHTS RESERVED
|